Alpha-diimines
The simple α-diimine (alpha-diimine) formed a huge part of my PhD thesis and is a molecular motif which I still believe to be extremely powerful. I include some of the material from my thesis here in draft form whilst I build a more general level introduction. Consider this a draft for now until I add a few more structures and pretty pictures.
So what is an α-diimine?
In short it is two imine moieties bonded at the α-carbon forming a motif observed in the generic scheme below. An imine being a nitrogen double carbon (N=C) function so a diimine becomes N=C-C=N.
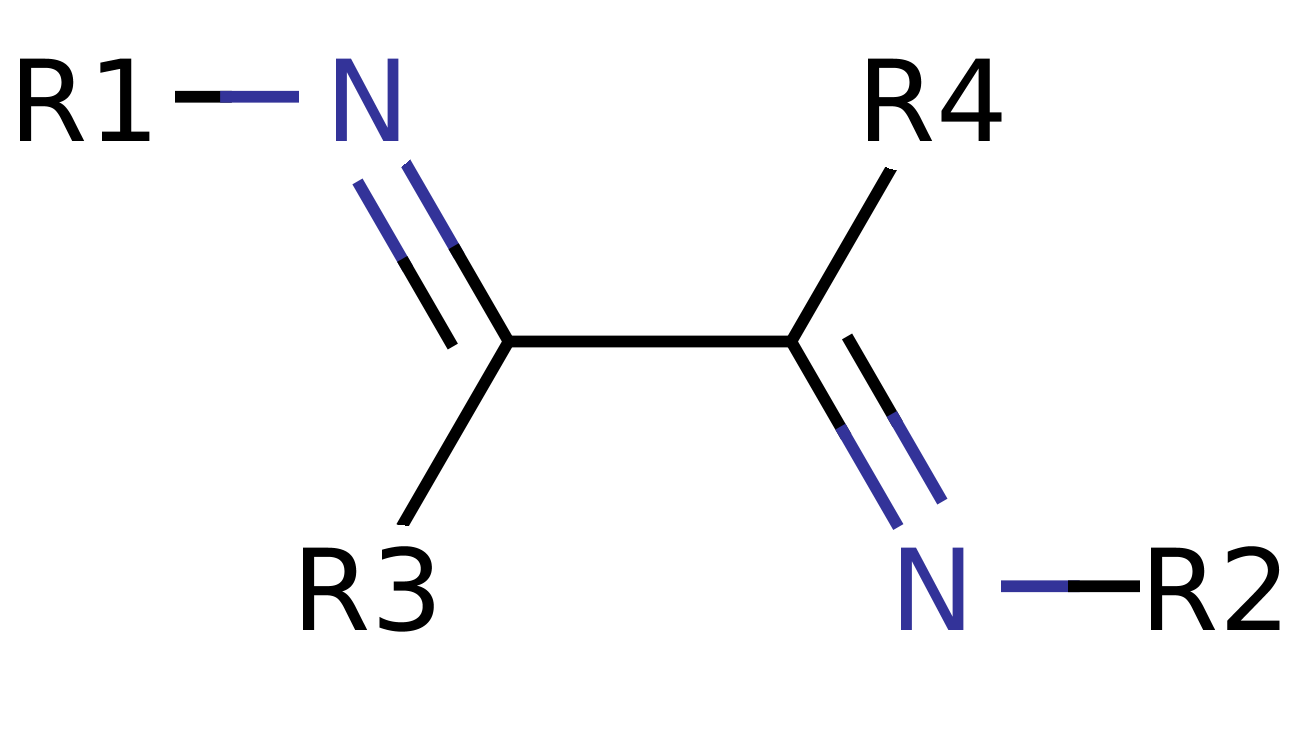
The coordination and molecular chemistry of the imine moiety and more specifically the α-diimine has seen substantial interest over the past thirty years. The ligand has both flexibility in coordination mode and electronic properties. Coupled together this provides for interesting and diverse chemistry.
General Background
α-diimine ligands are assuming increasingly important roles in catalytic and structural chemistry. Kees Vrieze and Gerard van Koten published a substantial review in 1983 surveying the wealth of α-diimine chemistry up until that date 1. The following introduction will therefore offer a brief summary of α-diimine structural features synthesis, application and work into the synthesis of asymmetric α-diimines will be introduced separately at somepoint.
The α-diimine moiety, or 1,4-diaza-1,3-butadiene (DAB), contains a N=C-C=N conjugated backbone which allows for substitution at the 2, 3- or a-carbon positions (denoted by a suffix to the DAB formula) or modification of the imine nitrogen R-group (denoted be a prefix to the DAB formula). The result of these simple alterations to the N=C-C=N backbone allows for dramatic changes in the steric and electronic environment of the N=C-C=N moiety. Both bipyridyl (bipy) and 1,10-phenanthroline (phen) show structural features similar to the α-diimine.
Benedix and co-workers report a series of NDDO calculations for the LUMO energy of selected α-diimines, in the s-cis (the terms s-cis and s-trans refer to the torsional isomers around the central C-C carbon bond of the 1,3-diene skeleton) configuration around the C-C bond to best replicate the geometry of coordinate bonding2. Their results suggest that the π-acceptor ability decreases in the order o-benzoquinonediimine > phen > Ph2DABH2 > (CH3)2DABH2 > (CH3)2DAB(Me)H > (CH3)2DABMe2 > 2,2’-bipyridine. They also suggest that 2,3-disubstitution with the phenyl- functionality, e.g. benzil, would not increase the π-acceptor capabilities of the ligand. These results are further substantiated by the change in the mean C-C bond length from 1.507(4) to 1.436(4) Å upon coordination.

The application of frontier orbital theory to the α-diimine N=C-C=N backbone helps reconcile the observed shortening of the C-C bond with π-acceptor behaviour. Donation of electron density from a low oxidation transition metal would be to the LUMO which would see an increase in the anti-bonding character between the C=N double bond causing elongation. Simultaneously an increase towards double bond character of the C-C single bond causes a decrease in bond length. The similarity between the central C-C distances in (c-hexyl)2DABH2, (1.46 Å) and in 1,3-butadienes (1.45 Å) underlies the isoelectronic relationship of the two classes of diene, and confirms the bond to be C(sp2)-C(sp2) in nature.
The rotation energy barrier between the s-trans and s-cis conformations has been determined by Benedix et al to be 22 kJ mol-1 giving a difference of 12.5 kJ mol-1 between the two conformational states 2.
Interestingly the solid state structure of compounds with increased steric bulk at the α-carbon position results in the formation of the s-cis conformation, i.e. where the N=C-C=N torsion angle is 90° and the ligand is therefore predisposed in a coordinating geometry and no rotational energy barrier needs to be overcome.
Kliegman and Barnes reported that the reaction of α-diimines with perchloric acid showed3,4 monobasic behaviour where the imine nitrogen R-group was not sterically demanding. The formation of a isolable five membered, highly stabilised, planar ring system is reported for (c-hexyl)2DABH2. This is only possible if the N=C-C=N skeleton can assume the E-s-cis-E conformation.
Johnson and co-workers have reported the acid-catalysed Z/E isomerisation of imines 5. Their findings suggest that there are two main mechanisms: 1) protonation-rotation via an iminium ion species and 2) nucleophilic catalysis. For the protonation-rotation mechanism, computational results would suggest that increased stabilisation of the iminium ion from donor groups of the imine helps to reduce the barrier to rotation, calculated at 77 kcal mol-1 for CH2=NH2 +. Nucleophilic catalysis requires the imine to undergo nucleophilic attack by the acid counter-ion giving a tetrahedral intermediate which can then undergo stereomutation by rotation and proton-exchange with subsequent loss of the nucleophile forming the other stereoisomer.
Due to the flexibility of the N=C-C=N skeleton and potential binding modes of the imine function whether it be via σ-N=C, π-N=C or both, α-diimines can potentially act in a variety of coordination modes, see table. The bonding modes involve not only the lone pairs of the N-atoms but also the π-C=N bonds and in some cases a five membered chelate ring similar to cyclopentadienyl.
Table: Bonding Modes for Range of α-diimine Complexes and the Number of Electrons Donated.
| Bonding Mode | Number of Electrons Donated | Compound |
| Monodentate | σ-N; 2 e- | Trans-PdCl2(PPh3)(_t-_Bu)2DABH2 |
| Bridging | σ-N, σ-N’; 2 e- + 2 e- | Trans-(PtCl2PBu3)2(_t-_Bu)2DABH2 |
| Chelating | σ,σ-N,N’ 4 e- | Mo(CO)4(_i_-Pr)2DABH2 |
| Bridging | σ-N, μ2-N’, η2-CN’ 6 e- | Fe2(CO)6 (_c_-hexyl)2DABH2 |
| Bridging | σ,σ-N,N’, μ2-CN, μ2-CN’ 8 e- | Mn2(CO)6(Me)2DABMe2 |
References
- G. V. Koten and K. Vrieze, 1, 4-Diaza-1, 3-butadiene (α-diimine) ligands: their coordination modes and the reactivity of their metal complexes, Advances in Organometallic Chemistry, 1982, 21, 151–239.
- J. Reinhold, R. Benedix, P. Birner, and H. Hennig, Quantum chemical investigations of the π-acceptor ability of α-diimine ligands, Inorganica Chimica Acta, 1979, 33, 209–213.
- J. M. Kliegman and R. K. Barnes, The conformation and NMR of conjugated diimines, Tetrahedron Letters, 1969, 10, 1953–1956.
- J. M. Kliegman and R. K. Barnes, Glyoxal derivatives—I: Conjugated aliphatic diimines from glyoxal and aliphatic primary amines, Tetrahedron, 1970, 26, 2555–2560.
- J. E. Johnson, N. M. Morales, A. M. Gorczyca, D. D. Dolliver, and M. A. McAllister, Mechanisms of Acid-Catalyzed Z/E Isomerization of Imines, The Journal of Organic Chemistry, 2001, 66, 7979–7985.